Saúde
Estudo sugere nova estratégia molecular para tratamento da síndrome do X frágil
O aumento da atividade de um componente específico dos receptores “NMDA” dos neurônios normalizou a síntese de proteínas, a atividade neural e a suscetibilidade a convulsões no hipocampo...
Por
David Orenstein - 05/03/2025


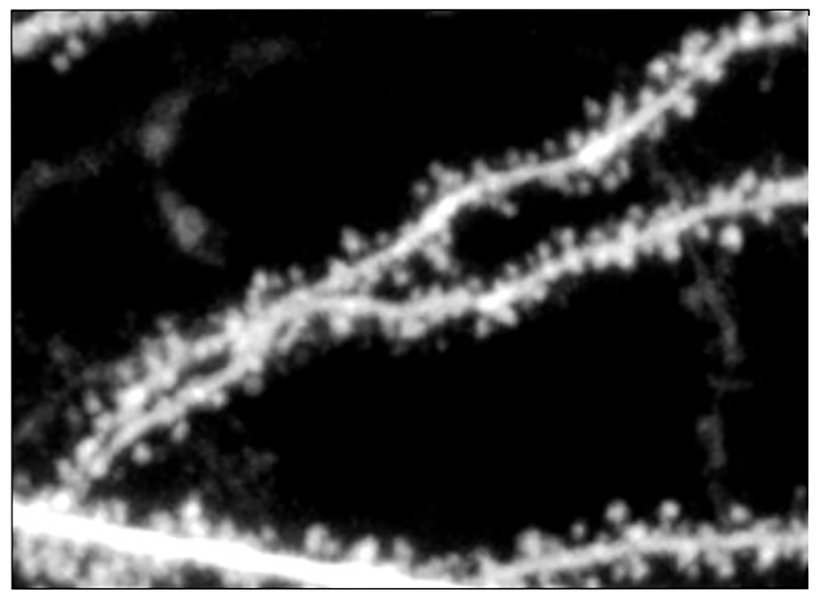
Observações das pequenas saliências que revestem os dendritos dos neurônios, chamadas espinhos, forneceram uma leitura crítica da função dos receptores NMDA das células no novo estudo, bem como em um precursor da pesquisa de 2020. Esta é uma imagem de microscópio de dois fótons, que está se aproximando dos limites da imagem óptica (daí seu desfoque). Créditos: Imagem: Stepanie Barnes/MIT Bear Lab
O aumento da atividade de um componente específico dos receptores “NMDA” dos neurônios normalizou a síntese de proteínas, a atividade neural e a suscetibilidade a convulsões no hipocampo de camundongos de laboratório com cromossomo X frágil.
Com base em mais de duas décadas de pesquisa, um estudo de neurocientistas do MIT no The Picower Institute for Learning and Memory relata uma nova maneira de tratar patologias e sintomas da síndrome do X frágil, o transtorno do espectro autista mais comum, causado geneticamente. A equipe mostrou que aumentar um novo tipo de sinalização de neurotransmissor reduziu as características do X frágil em modelos de camundongos do transtorno.
A nova abordagem, descrita em Cell Reports , funciona mirando uma subunidade molecular específica de receptores “NMDA” que eles descobriram que desempenha um papel fundamental em como os neurônios sintetizam proteínas para regular suas conexões, ou “sinapses”, com outros neurônios em circuitos cerebrais. Os cientistas mostraram que em camundongos modelo X frágil, aumentar a atividade do receptor fez com que os neurônios na região do hipocampo do cérebro aumentassem a sinalização molecular que suprimiu a síntese excessiva de proteína em massa, levando a outras melhorias importantes.
Arrumando a mesa
“Uma das coisas que acho mais satisfatórias sobre este estudo é que as peças do quebra-cabeça se encaixam tão bem no que veio antes”, diz o autor sênior do estudo Mark Bear , professor Picower no Departamento de Ciências Cognitivas e do Cérebro do MIT. A ex-pós-doutoranda Stephanie Barnes, agora palestrante na Universidade de Glasgow, é a autora principal do estudo.
O laboratório de Bear estuda como os neurônios editam continuamente suas conexões de circuito, um processo chamado de "plasticidade sináptica" que os cientistas acreditam ser a base da capacidade do cérebro de se adaptar à experiência e de formar e processar memórias. Esses estudos levaram a duas descobertas que prepararam o terreno para o avanço recém-publicado. Em 2011, o laboratório de Bear mostrou que o X frágil e outro transtorno do autismo, a esclerose tuberosa (Tsc), representavam duas extremidades de um continuum de um tipo de síntese de proteína nos mesmos neurônios. No X frágil havia muito. No Tsc havia muito pouco. Quando os membros do laboratório cruzaram camundongos X frágil e Tsc, de fato, seus descendentes emergiram saudáveis, pois as mutações de cada transtorno essencialmente se cancelaram.
Mais recentemente, o laboratório de Bear mostrou uma dicotomia diferente. Há muito tempo se entende, a partir de seu trabalho influente na década de 1990, que o fluxo de íons de cálcio através dos receptores NMDA pode desencadear uma forma de plasticidade sináptica chamada “depressão de longo prazo” (LTD). Mas em 2020, eles descobriram que outro modo de sinalização pelo receptor — um que não exigia fluxo de íons — alterava a síntese de proteínas no neurônio e causava um encolhimento físico das estruturas dendríticas de “espinha” que abrigam sinapses.
Para Bear e Barnes, esses estudos levantaram a perspectiva de que se eles pudessem identificar como os receptores NMDA afetam a síntese de proteínas, eles poderiam identificar um novo mecanismo que poderia ser manipulado terapeuticamente para tratar a patologia e os sintomas do X frágil (e talvez esclerose tuberosa). Isso seria um avanço importante para complementar o trabalho em andamento que o laboratório de Bear fez para corrigir os níveis de síntese de proteína do X frágil por meio de outro receptor chamado mGluR5.
Dissecção do receptor
No novo estudo, a equipe de Bear e Barnes decidiu usar o efeito não iônico no encolhimento da espinha como uma leitura para dissecar como os NMDARs sinalizam a síntese de proteínas para plasticidade sináptica em neurônios do hipocampo. Eles levantaram a hipótese de que a dicotomia de efeitos iônicos na função sináptica e efeitos não iônicos na estrutura da espinha pode derivar da presença de dois componentes distintos de receptores NMDA: "subunidades" chamadas GluN2A e GluN2B. Para testar isso, eles usaram manipulações genéticas para eliminar cada uma das subunidades. Quando fizeram isso, descobriram que eliminar "2A" ou "2B" poderia eliminar LTD, mas que apenas eliminar 2B afetava o tamanho da espinha. Experimentos posteriores esclareceram que 2A e 2B são necessários para LTD, mas que o encolhimento da espinha depende apenas da subunidade 2B.
A próxima tarefa era resolver como a subunidade 2B sinaliza o encolhimento da espinha. Uma possibilidade promissora era uma parte da subunidade chamada de “domínio carboxiterminal”, ou CTD. Então, em um novo experimento, Bear e Barnes aproveitaram um camundongo que havia sido geneticamente modificado por pesquisadores da Universidade de Edimburgo para que os CTDs 2A e 2B pudessem ser trocados um pelo outro. Um resultado revelador foi que quando a subunidade 2B não tinha seu CTD adequado, o efeito na estrutura da espinha desaparecia. O resultado afirmou que a subunidade 2B sinaliza o encolhimento da espinha por meio de seu CTD.
Outra consequência da substituição do CTD da subunidade 2B foi um aumento na síntese de proteínas em massa que se assemelhava aos achados no X frágil. Por outro lado, o aumento da sinalização não iônica por meio da subunidade 2B suprimiu a síntese de proteínas em massa, lembrando o Tsc.
Tratando X frágil
Juntando as peças, as descobertas indicaram que aumentar a sinalização por meio da subunidade 2B poderia, assim como introduzir a mutação que causa o Tsc, resgatar aspectos do X frágil.
De fato, quando os cientistas trocaram a subunidade 2B CTD do receptor NMDA em camundongos modelo X frágil, eles encontraram correção não apenas da síntese excessiva de proteína em massa, mas também plasticidade sináptica alterada e excitabilidade elétrica aumentada que são marcas registradas da doença. Para ver se um tratamento que tem como alvo os receptores NMDA pode ser eficaz no X frágil, eles tentaram um medicamento experimental chamado Glyx-13. Este medicamento se liga à subunidade 2B dos receptores NMDA para aumentar a sinalização. Os pesquisadores descobriram que este tratamento também pode normalizar a síntese de proteína e reduzir convulsões induzidas por som nos camundongos X frágil.
A equipe agora levanta a hipótese, com base em outro estudo anterior em laboratório, de que o efeito benéfico da sinalização CTD da subunidade 2B em camundongos com cromossomo X frágil é que ela desloca o equilíbrio da síntese de proteínas de uma tradução muito eficiente de RNAs mensageiros curtos (o que leva à síntese excessiva de proteínas em massa) para uma tradução de menor eficiência de RNAs mensageiros mais longos.
Bear diz que não sabe quais são as perspectivas do Glyx-13 como um medicamento clínico, mas observou que há alguns medicamentos em desenvolvimento clínico que têm como alvo específico a subunidade 2B dos receptores NMDA.
Além de Bear e Barnes, os outros autores do estudo são Aurore Thomazeau, Peter Finnie, Max Heinreich, Arnold Heynen, Noboru Komiyama, Seth Grant, Frank Menniti e Emily Osterweil.
A Fundação FRAXA, o Instituto Picower para Aprendizagem e Memória, a Fundação Freedom Together e os Institutos Nacionais de Saúde financiaram o estudo.